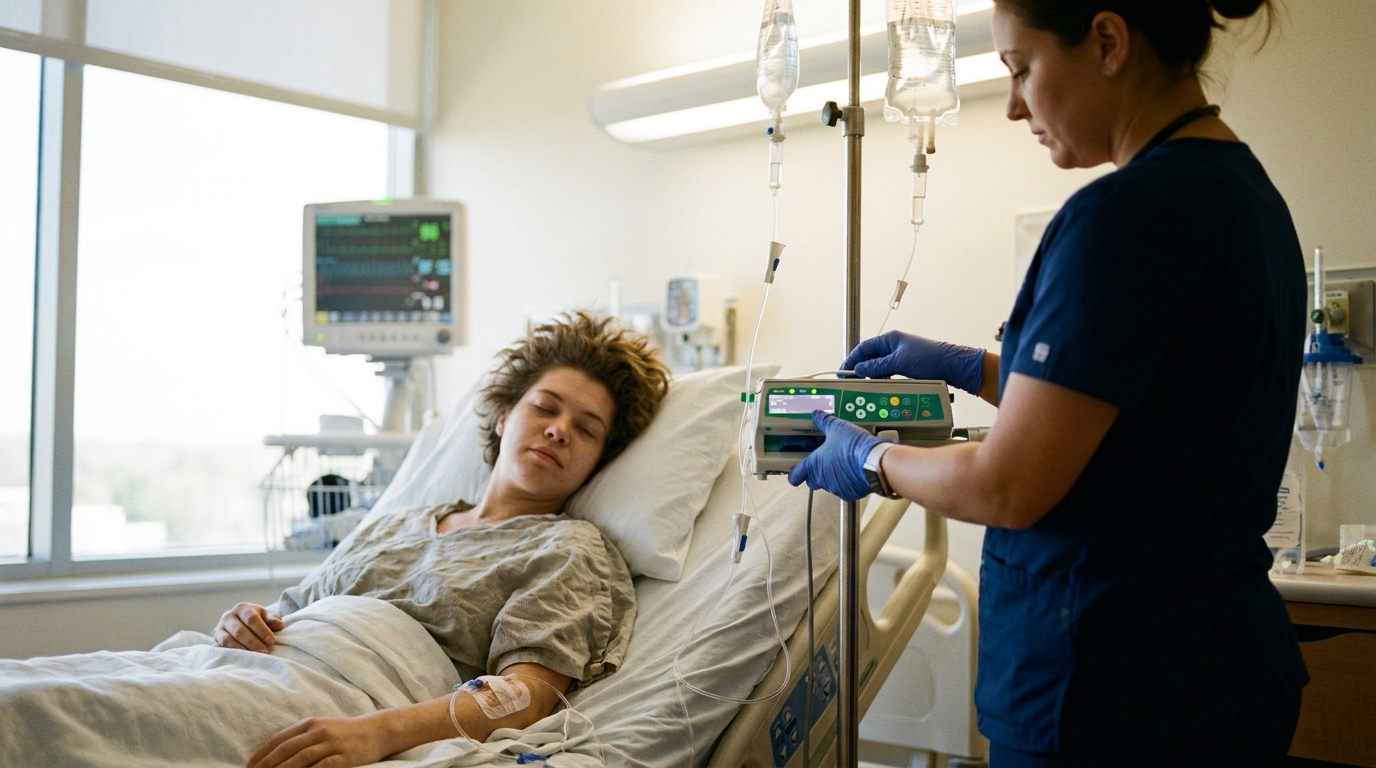
Base Editing of Gamma-Globin Promoters Increases Fetal Hemoglobin in Sickle Cell Disease
Interim phase 1-2 data show risto-cel achieves 60% fetal hemoglobin and eliminates severe vaso-occlusive crises in 31 patients.
The Clinical Lighthouse
Already a member?
or
The Clinical Lighthouse
Subscribe to read the full analysis
Full access to every article, clinical summary, and the daily evidence brief.
- Unlimited access to all articles
- Daily evidence digest
- Daily podcast with news summaries
- Lighthouse Guideline Briefings
- Ask Lumi: your guide to search, summarise, and explain the archive
- Cancel anytime