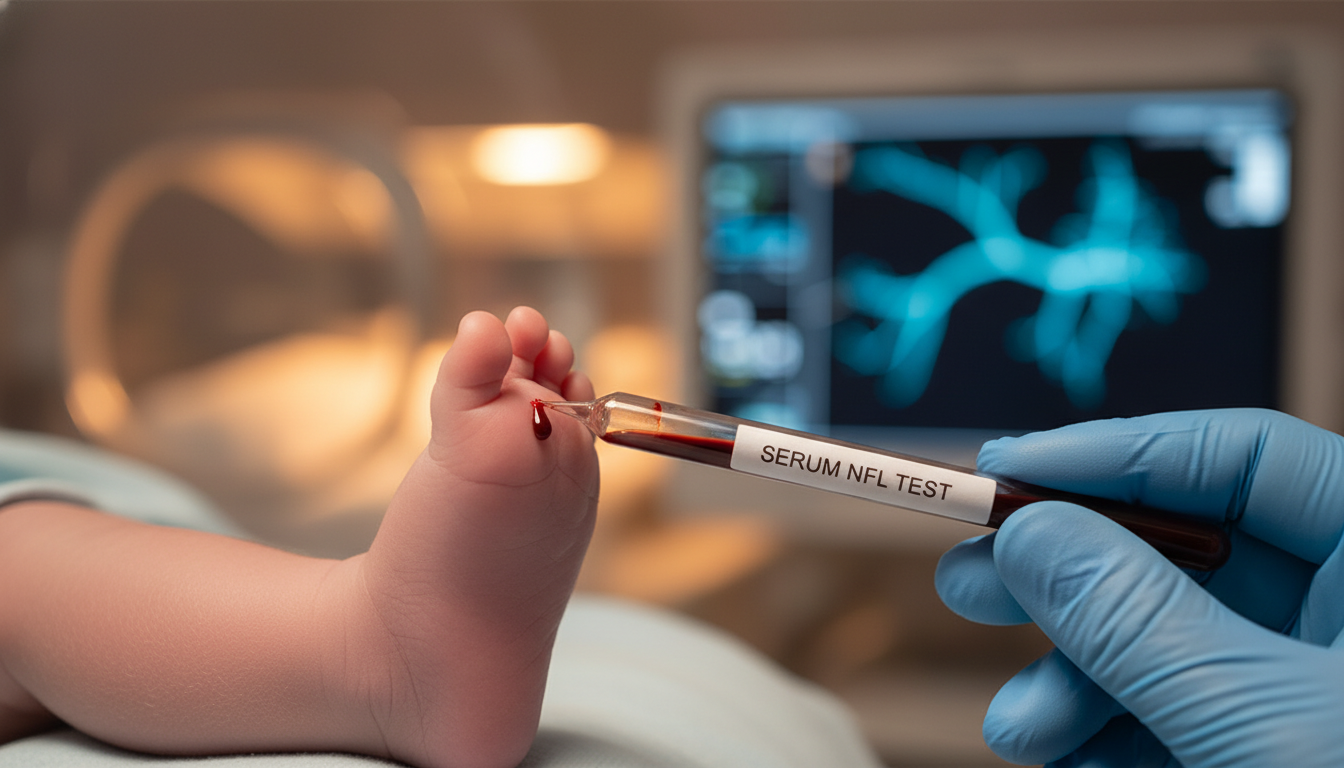
Serum Neurofilament Light Chain Identifies Subclinical Axonal Injury in SMA Neonates
Elevated serum neurofilament light chain levels predict two-year motor outcomes and reveal active denervation before clinical symptoms appear.
The Clinical Lighthouse
Already a member?
or
The Clinical Lighthouse
Subscribe to read the full analysis
Full access to every article, clinical summary, and the daily evidence brief.
- Unlimited access to all articles
- Daily evidence digest
- Daily podcast with news summaries
- Lighthouse Guideline Briefings
- Ask Lumi: your guide to search, summarise, and explain the archive
- Cancel anytime